Abstract
Aging mammalian skeletal muscle satellite cells (MuSCs) undergo a decline of stem cell/progenitor cell proliferative and regenerative capacity, and the development of a physiological milieu characteristic of a state of chronic sterile inflammation. p38αMAPK and ERK1/2 are two major signaling pathways that regulate the age-associated decline of MuSC proliferative capacity. In this review we propose the following mechanism that links the p38αMAPK pathway to the decline of self-renewal and regenerative capacity of aged MuSCs: a) the HS-FGF-2-FGFR1-p38αMAPK-Axis, a tightly linked homeostatic signaling complex, is in synchrony with the autoinhibition of FGFR1; b) autoinhibition contributes to the Axis’ regulation of the homeostasis of P-p38αMAPK activity in juvenile MuSC; c) this combination of protein-protein interactions is characteristic of a juvenile cytoplasmic milieu of beneficial P-p38αMAPK activity and d) includes Sprouty1 inhibition that supports the stimulation of FGF-2 --> miR-29a; e) the miR29a dismantles the basement membrane in preparation for the initiation of replication; f) an age-associated impaired, dysregulated, over-sulfated heparan sulfate ligand (HS)-FGF-2 fails to activate FGFR1 in aged MuSCs; g) this uncouples its regulation of p38αMAPK and ERK1/2 pathways and results in desensitization of FGFR1; h) desensitization of FGFR1 and Sprouty1 interaction in aged MuSC uncouples their regulation of P-p38αMAPK in the aged MuSCs; i) this enables a state of chronic sterile inflammation to promote and sustain an increased level of P-p38αMAPK activity; and, j) the increased activity of P-p38αMAPK in aged MuSC stimulates the production of cell cycle inhibitors, miR-1 and miR-133, thereby attenuating the expression of the cell cycle regulators, SP1 and cyclin D1, resulting in a G1/S arrest; j) the increased level of p38αMAPK activity promotes the apoptosis of the aged activated MuSCs.
This mechanism involves the synergistic interactions of HS-FGF2-FGFR-1, Sprouty (spry1), miR-1, miR-133 and miR-29a that unify the extracellular niche and intracellular milieu for the juvenile vs age-associated regulation of proliferative capacity of the MuSC. Our hypothesis unifies these interactions with the role of the extracellular niche and intracellular milieu in the stimulation of juvenile proliferation vs age-associated decline of skeletal muscle satellite cell self-renewal and regenerative proliferation. Word Count = 344
Author Contributions
Academic Editor: Jean-Francois Grosset, University of Technology of Compiegne, France.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2020 John Papaconstantinou
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Background
Overview of the Role of the HS-FGF-2-FGFR1-p38αMAPK Axis, p38αMAPK and Sprouty 1 in the Regulation of Aged Skeletal Muscle Satellite cell Proliferation.
Aging mammalian skeletal muscle satellite cells (MuSC) progress simultaneously with the development of a physiological milieu characteristic of a state of chronic inflammation. It is accompanied by what is referred to as an “irreversible growth arrest (IGA)” – a decline of stem cell/progenitor cell self-renewal and regenerative capacity 1, 2, 3, 4, 5, 6, 7, 8. This age-associated loss of MuSC replicative capacity is, however, reversible by the inhibition of p38α/βMAPK activity 1, 2 or exposure, by parabiosis, to a young physiological environment 9, 10, 11, 12. In this proposal, we refer to aged MuSCs as senescent cell quiescence (SCQ), i.e. senescent cells that have retained a reversible replicative capacity 13. This characteristic of aging stem and progenitor cells occurs in multiple tissues including heart muscle 14, hematopoietic 15, lung 16, neuronal 17, pancreatic β cells 18, 19, and hepatocytes 20.
There are multiple signaling processes such as p38αMAPK, ERK1/2 and NFkB [21] and factors such as syndecan 4, heparan sulfate, β-integrin, fibronectin, etc., involved in the regulation of skeletal MuSC proliferation, 3,22. Our model specifically focuses on the role of heparan sulfate (HS) in the (HS)-FGF-2-FGFR1-p38αMAPK Axis signaling pathway and Sprouty1 (a tyrosine kinase inhibitor of the FGFR1 kinase domain) that regulate juvenile MuSC proliferative capacity. We propose a mechanism that links the desensitization of aged skeletal MuSC FGFR1 signaling to an impaired response by the receptor due to over-sulfated heparan sulfate-FGF-2 (HS-FGF-2) ligand [1, 2, 4, 22, 23]. Dysregulation of the HS-FGF-2-FGFR1-p38αMAPK-Axis signaling capacity results in an age-associated decline of MuSC self-renewal and proliferative capacity and the development of SCQ (Figure 1) 13
Figure 1.The role of the HS-FGF-2–FGFR1-p38αMAPK Axis in the regulation of skeletal muscle satellite cell (MuSC) proliferation-differentiation in (A) juvenile (young) and (B) aged MuSCs. (A) Juvenile (Young) MuSC: The level of P-p38αMAPK activity in juvenile MuSC is regulated by FGFR1 autoinhibition and is coupled to and regulated by the interaction of HS-FGF-2 with FGFR1 that forms and activates the HS-FGF2-FGFR1-p38αMAPK-Axis. This regulates the beneficial homeostatic level of p38αMAPK activity and downregulates miR-1/miR-133 synthesis thus stimulating the expression of the cell cycle regulators, Sp1 and cyclin D1 and the progression of myoblast proliferation. (B) Aged – Senescent MuSC: Control of P-p38αMAPK is uncoupled by the desensitization-dysregulation of FGFR1 signaling; its level of activity is thus increased in response to elevated endogenous sterile inflammation in the absence of FGFR1 involvement. The increased P-p38αMAPK activity stimulates miR-1/miR-133 levels thereby enhancing their inhibition of Sp1 and Cyclin D1 respectively; this results in the G1/S cell cycle arrest. (C). Reversal of IGA in Aged Tissue: Reversal of the Cell Cycle arrest in aged skeletal MuSC is as follows: The age-associated over-sulfation of HS decreases, i.e., it is down regulated to the levels in juvenile MuSC. The HS-FGF-2 interaction with FGFR1 is reinitiated thereby establishing the level of juvenile FGFR1-coupled regulation of MuSC P-p38αMAPK; the decline of miR-1 and miR-133 levels, promotes reactivation of Sp1 and Cyclin D1; cell cycle progression and cell proliferation is initiated, i.e., reprogramming of the aged MuSCs. This Figure does not include the synergistic interaction of spry1 with the kinase domain.
Homeostatic intercommunications between MuSCs, the muscle milieu and microenvironment, control the normal growth and self-renewal of juvenile (young adult) skeletal muscle. We will discuss the alterations of these intercommunications that play an important role in the regulation of muscle regeneration and the pathophysiological consequences of aging on these functions 4.
It is important to recognize that uncontrolled FGF-ligand signaling is associated with malignancies and other pathophysiological syndromes thus requiring a self-regulatory control of physiological protective mechanisms 24. This is accomplished by FGFR1 autoinhibition, a process that serves as a self - defense mechanism that minimizes inadvertent FGFR1 signaling. Autoinhibition of FGFR1 thus also serves as a strongly coupled regulator of beneficial p38αMAPK activity in juvenile skeletal muscle 24. Our model thus proposes that the HS-FGF-2-FGFR1-p38αMAPK Axis is a molecular self-defense system that protects against inadvertent FGFR1 signaling thereby regulating the beneficial level of FGFR1 signaling and consequently, the beneficial level of activity of its p38αMAPK target. Furthermore, as aging progresses the juvenile MuSCs develop a desensitization-dysregulation of the HS-FGF-2-FGFR1 signaling interaction [1, 2] that enables the development of an elevated, sustained state of chronic inflammation, a consequence of endogenous-sterile inflammation. This promotes an elevated sustained level of P-p38αMAPK activity that is independent of MuSC FGFR1 signaling [25,26,27,28]. We thus propose that the tightly linked HS-FGF-2-FGFR1-p38α MAPK signaling complex regulates and maintains the beneficial level of P-p38αMAPK activity in juvenile MuSCs by controlling the level of autoinhibition of FGFR1; this autoinhibition is uncoupled in the aged satellite cells thereby enabling the physiological state of chronic sterile inflammation to promote and sustain an elevated P-p38αMAPK activity.
As tissues age they develop a state of chronic inflammation characterized by the elevated, sustained activity of P-p38α/βMAPK 1, 2, 23, 29 and, more specifically p38αMAPK 18, 23, 30. Furthermore, p38αMAPK plays a key role in the regulation of MuSC functions associated with regenerative capacity and the decline of self-renewal 1, 2, 23, 30, 31, their exit from the cell cycle, the progression of their terminal differentiation to form myofibers, and the decline of MuSC pool levels 1, 2, 4, 32.
The decline of proliferative capacity of senescent MuSCs is linked to increased levels of over-sulfated heparan sulfate-FGF-2-ligand (HS-FGF-2) and the desensitization - dysregulation of a potentially over-sulfated HS-fibroblast growth factor receptor-1 (HS-FGFR1). This results in the loss of control of p38α/βMAPK activity 13, 30, 30, 31, 32, 33, 34, 35, 36, 37, 38which is linked to the inability of desensitized HS-FGFR1 to respond to HS-FGF-2 ligand. This enables the sterile chronic inflammation to promote an uncontrolled elevation of P-p38αMAPK activity and the decline of stem cell/progenitor cell proliferative and regenerative capacity. This is a physiological characteristic of inflammation that is linked to the loss of MuSC asymmetric replication 1, 2, 3, 4. These characteristics are major components of the aging skeletal muscle phenotype. Importantly, a consequence of the increased P-p38αMAPK activity results in the increased transcription of the cell cycle inhibitors, miR-1/miR-13338, 39; these miRNAs attenuate the expression of the cell cycle regulators, SP1 and cyclin D1, resulting in a G1/S arrest (Figure 1). Thus, the upregulation of HS-FGF-2 that occurs in juvenile muscle regeneration stimulates the production of Sp1 and cyclin D2 and myoblast proliferation at the early developmental stage of regeneration when the level of P-p38αMAPK activity is controlled by the HS-FGF-2-FGFR1-p38αMAPK Axis. We thus propose that the age-associated stimulation of transcription of the cell cycle-inhibiting miRNAs, miR-1 and miR-133, that directly control the promotion of MuSCs SCQ, is increased by the elevated P-p38αMAPK activity. Meanwhile, the desensitization-dysregulation of the HS-FGF-2-FGFR1 interaction uncouples its coordinated and rigorous control of P-p38αMAPK activity. This allows the chronic inflammation-mediated elevation of P-p38αMAPK activity and miRNA transcription. Consequently, the dysregulation of the HS-FGF-2-FGFR1-p38αMAPK Axis and the elevated P-p38αMAPK activity associated with chronic sterile inflammation promotes the development of the SCQ in aged tissues.
Skeletal muscle extracellular matrix plays a critical role in the maintenance of life-long ability of MuSCs to sustain their stem cell functions. This is attributed to their ability to maintain the reversibility of their quiescence in response to muscle injury. These stem cells maintain self-renewal and differentiation via asymmetric replication 40.
MuSCs perform asymmetric replication to produce a replicatively competent daughter cell while the other cell proceeds to terminal differentiation. At this time a subpopulation of cells returns to replenish the satellite cell pool and quiescence. This physiological cycle, which is maintained throughout life is programmed for activation and return to dormancy as a critical function for muscle self-renewal and injury repair. This physiological cycle is dependent upon extracellular matrix regulators of stem cell quiescence within the niche-intracellular signaling interaction that promotes quiescence. The development, therefore, of a reversible quiescent state is mediated by Sprouty1 (spry 1) a receptor tyrosine kinase (RTK) inhibitor, and miR-29a, an inhibitor of basement membrane (BM) proteins that enables dismantling of the BM and the initiation of proliferation. Furthermore, we propose that these inhibitors work synergistically with the FGF-2-FGFR1-p38αMAPK-Axis to achieve their contribution to the reversibility of MuSC quiescence in the juvenile muscle. For example, the juvenile quiescent MuSCs break out of quiescence in response to muscle injury under controlled levels of P-p38αMAPK and low levels of over-sulfated HS-FGF-2. These physiological conditions support the progression of myofiber (myogenesis) formation. On the other hand, in the aged muscle, the cells that break out of quiescence do so under physiological conditions of elevated over-sulfated FGF-2, desensitized FGFR1 and elevated levels of P-p38αMAPK activity. The latter, elevated levels of p38αMAPK, strongly supports the pathway to apoptosis, which would explain the observation that these cells do not participate in myogenesis.
Main Text
The Role of p38αMAPK in the Regulation of Skeletal MuSC Self-Renewal and Regenerative Capacity.
Our model proposes that a) age-associated chronic sterile inflammation and over-sulfated heparan drive the desensitization of HS-FGF-2-FGFR1 signaling that, b) uncouples the level of p38αMAPK activity thereby allowing increased levels of p38αMAPK activity and the c) loss of asymmetric distribution of P-p38α and attenuation of their replication, but permitting their differentiation 1. The uncoupling of the HS-FGF-2-FGFR1-p38αMAPK Axis thus allows the activity of P-p38αMAPK to continue independent of its interaction with and control by FGFR1, thereby maintaining the production of the cell cycle inhibiting miRNAs and the G1/S arrest. Furthermore, as the HS-FGF-2-FGFR1 signaling regulates the levels of P-p38αMAPK activity in juvenile MuSCs, the consequences of aging on this regulation dictate the loss of proliferative activity and the gain of quiescence of the MuSC. The replicative capacity of aged MuSC is thus no longer controlled by HS-FGF-2-FGFR1 signaling but is taken over by the chronic inflammation-mediated levels of P-p38αMAPK activity. Restoration of self-renewal and regenerative capacity must therefore be achieved by the reprogramming of the HS-FGF-2-FGFR1-p38αMAPK-Axis activity to its level of coupled and coordinated juvenile homeostasis. That this reprogramming of aged cells can be achieved has been experimentally shown by specific small molecule inhibition of p38αMAPK activity or parabiosis and grafting1, 2, 3, 4, 1, 12
p38αMAPK Regulates Myoblast Differentiation and Cell Cycle Exit in aged Skeletal MuSC
p38αMAPK signaling plays a key role in the regulation of expression of skeletal muscle genes that control myotube formation3, 30, 41. Myoblasts deficient in p38αMAPK are primarily uninuclear and exhibit a severe defect in their ability to form multinuclear myotubes whereas cells deficient in p38β, p38γ or p38δ display a multinucleated morphology (Figure 2)30, 41. This suggests that p38αMAPK plays an important role in the regulation of myogenesis 30, 41. The defective myogenesis attributed to p38αMAPK deficiency is the basis for the delayed cell cycle exit, and continuous proliferation 30, 41. The p38αMAPK genetic deficiency experiments suggest that its inhibition by small molecule inhibitors should result in the failure to initiate terminal differentiation i.e., failure to exit the cell cycle and to produce myotubes (Figure 2). We thus propose that the elevated level of P-p38αMAPK activity plays a key role in the development of SCQ. Furthermore, the loss of asymmetric distribution of P-p38αMAPK to the daughter cells promotes the loss of asymmetric proliferation suggesting that the continuation of terminal differentiation by both daughter cells may account for the decrease of the MuSC pool level.
Figure 2.Multinucleated myotube formation is inhibited by p38αMAPK deficiency. Wt and p38αD/DMAPK myoblast cultures were induced to undergo myoblast fusion. The immunohistochemical data show that myogenic cells do not exhibit cell cycle withdrawal and terminal differentiation; that p38αMAPK deficient myoblasts have impaired ability to exit the cell cycle; the p38aD/DMAPK myoblasts also exhibit an enhanced proliferative potential. Myogenic cells lacking p38αMAPK possess an increased propensity for self-renewal. (This figure is a modification of Figure 3A from Perdiguera et al. EMBO J 26, 1245, (2007) with permission of the publisher.)
Our model suggests that partial inhibition, genetically or pharmacologically, of p38αMAPK in aged mice should either restore 1, 2, or delay the loss of the self-renewal of aged MuSC [18, 22, 23]. Based on studies using inhibitors of p38α/β we demonstrated that the partial and specific inhibition of p38αMAPK achieved by these inhibitors could be achieved in vivo by a dominant negative mutation of the p38αMAPK allele (DN-p38αAF/+) and that this mutant would delay the decline of proliferative capacity 18, 22, 23. Using this mutant we demonstrated that the genetic attenuation of p38αMAPK in gastrocnemius MuSC, in vivo, delays the development of multiple age-associated markers that includes the delayed decline of BuBR1, the attenuated expression of p16Ink4a and p19Arf and the elevated protective activity of ALDH1A1, 22, 23.
Using primary myoblasts derived from neonatal mice deficient in p38α, p38β, p38γ, or p38δ MAPKs has shown that p38αMAPK is the signaling protein that is central to myogenesis in vitro and in vivo30, 41. Moreover, p38αMAPK was shown to promote muscle differentiation and fusion and to be a critical regulator of myoblast cell cycle exit which leads to activation of the muscle differentiation gene program 30, 41. Thus, with the increased level of P-p38αMAPK and the increased level of miRNA-1 and miRNA-133 the replicative capacity of the senescent MuSC is inhibited. In our model, the inactivation (desensitization) of FGFR1 in aged MuSC enables sterile inflammation to independently promote the elevation and maintenance of P-p38αMAPK activity and the production of cell cycle inhibiting miRNAs that result in G1/S arrest. An important characteristic of our model is that p38αMAPK activity is required for the downregulation of cell proliferation.
p38αMAPK Down-Regulates Cyclin D1
Increased cyclin D1 expression in p38αMAPK-deficient myoblasts supports their continued proliferation 30, 41. Inhibition of p38αMAPK, however, shown to attenuate myofibrogenesis and maintain proliferation of adult mammalian MuSC and cardiomyocytes supports our proposal for skeletal MuSC 30, 41. Furthermore, constitutive activation of p38αMAPK in human rhabdomyosarcoma cells restores growth arrest and terminal differentiation. For example, MyoD inhibits proliferation and promotes muscle differentiation but in rhabdomyosarcoma, the differentiation program is blocked, and proliferation is deregulated despite MyoD expression. p38αMAPK activation, which is essential for muscle differentiation, is deficient in rhabdomyosarcoma cells and the enforced induction by MKK6EE, which activates MKK6 --> p38αMAPK, restores MyoD function, growth arrest and terminal differentiation. These studies support the proposal that activation of P-p38αMAPK is required for myogenic differentiation 32.
miR-1 and miR-133 Control cell Cycle Progression of Myoblasts via Targeting of Cyclin D1, a key Regulator of G1/S Transition
The elevated levels of P-p38αMAPK activity in aged MuSCs attenuates cell cycle progression while promoting the development of SCQ (Figure 1;38, 39. This is mediated by the increased levels of miR-1 and miR-133 and their interactions with the 3’UTR (PolyA) of Sp1 (miR-1) and cyclin D1 (miR-133) mRNAs (Figure 1)38, 39. We propose a novel mechanism for the development and maintenance of the age-associated SCQ that is linked to, a) the level of expression of over-sulfated HS-FGF-2 and its failure to activate the FGFR1-p38αMAPK-Axis. This allows the elevation P-p38αMAPK activity and, b) increases levels of miRNA cell cycle inhibitors and, c) the elevated levels of inhibition of the tyrosine kinase domain of FGFR1 by Sprouty1.
An Alternative Model of the Regulation of MuSC Regenerative capacity
In this alternative model FGF-2 regulates juvenile skeletal MuSC regenerative capacity based on its ability to inhibit p38αMAPK signaling during the early phase of regeneration thereby lowering the levels of miR-1 and miR-133 synthesis and enabling the Cyclin D1- and Sp1-mediated progression of the cell cycle and myoblast proliferation (Figure 1) 38, 39. The model proposes that FGF-2 released from a frozen tissue myotrauma - promoted regeneration, represses p38αMAPK signaling thereby attenuating the expression of miR-1/miR-133.This facilitates satellite cell proliferation and the progression of regeneration. Importantly, this sequence of events reportedly occurs in the early phase of regeneration, i.e., the proliferation phase of the activation of muscle regeneration38, 39. This may also involve FGFR1 autoinhibition-mediated attenuation of p38αMAPK activity thereby decreasing miRNA levels. This inhibition regulates the levels of p38αMAPK that benefit regenerative proliferation. Furthermore, this is consistent with our proposal that the level of P-p38αMAPK is coupled to the level of autoinhibition of HS-FGF-2-FGFR1-p38αMAPK–Axis thus enabling a controlled homeostatic level of juvenile MuSC proliferation to proceed. The consequences of our model vs. the alternative model are otherwise similar in that the HS-FGF-2-FGFR1 mediated attenuation of p38αMAPK activity enables muscle differentiation and fusion to proceed as well as the regulation of cell cycle exit. Furthermore, as the down regulator of miR-1 and miR-133 it up regulates cyclin D1 mRNA expression by HS-FGF-2 thereby supporting proliferation These results also showed that the specific inhibition of FGFR1 with PD173074 blocked its signaling and attenuated the inhibitory effect of FGF-2 on the transcription of miR-1 and miR-133 in isolated gastrocnemius. These data suggest that HS-FGF-2-FGFR1 promoted the decline of miRNA via the inhibition of p38αMAPK activity. These studies conclude that in the early stages of regeneration, HS-FGF-2 interacts with FGFR1 to activate HS-FGF-2-FGFR1 signaling and that this attenuates the inhibition of the cell cycle and promotes the progression of MuSC proliferation. In this model, the role of FGF-2 in the regulation of juvenile MuSC renewal is based on its ability to attenuate the P-p38αMAPK signaling pathway during the early replicative phase of regeneration thereby lowering the levels of miR-1 and miR-133 synthesis and enabling the Cyclin D1- and Sp1-mediated progression of the cell cycle and myoblast proliferation [38]. The controlled level of FGF-2-P-p38αMAPK activity thus plays a key role in the induction of skeletal MuSC proliferation [38, 42]. For example, myoblasts with a deficiency of p38αMAPK exhibit delayed cell cycle exit and continuous proliferation in vitro, indicating that P-p38αMAPK is a critical regulator of myoblast exit [30, 41], which is a necessary step prior to commencing the muscle differentiation program [30]. Interestingly, upon differentiation to myofibers the P-p38αMAPK level increases. This is consistent with the loss of proliferative capacity by the terminally differentiated myoblasts. Furthermore, P-p38αMAPK signaling stimulates the transcription of miR-1 in hypoxic cardiac myocytes [39] and both miR-1 and miR-133 in regenerating rat liver [38, 39]. Thus, the p38αMAPK linked miR-1 and miR-133 are attenuated in muscle regeneration and up-regulated in quiescent aged cells; miR-1 and miR-133 thus induce myoblast growth arrest at the G1/S phase of the cell cycle.
The HS-FGF-2-FGFR1-p38αMAPK-Axis and Sprouty 1
The coupled coordinated signaling activities of the HS-FGF-2-FGFR1-p38αMAPK-Axis and Sprouty1 regulate specific processes of juvenile MuSC myogenesis. Chronic inflammation, desensitization-dysregulation of HS-FGF-2-FGFR1, the sustained elevated activity of the P-p38αMAPK, and the levels of inhibition of the kinase domain by Sprouty1 are major physiological characteristics of the aged skeletal muscle phenotype (Figure 1). These functions promote the loss of asymmetric distribution of P-p38αMAPK to daughter cells, self-renewal and regenerative proliferation 1, 2, 3. The mechanism underlying the age-associated decline of satellite cell replicative capacity involves the physiological desensitization-dysregulation of the HS-FGF-2-FGFR1-p38αMAPK-Axis signaling in combination with the elevated levels of Sprouty 1 (Figure 11, 2, 4. Furthermore, the HS-FGF-2-FGFR1- and Sprouty-linked, coordinated control of P-p38αMAPK activity in juvenile skeletal muscle enables the physiologically beneficial levels of P-p38αMAPK activity to perform its multiple functions that include the regulation of MuSC proliferation and differentiation. Development of insensitivity of the aged MuSC HS-FGF-2-FGFR1 signaling on the other hand, results in uncoupling of its regulation of P-p38αMAPK activity, while Sprouty inhibits the tyrosine kinase domain. This enables the sterile endogenous and/or exogenous inflammation-mediated level of P-p38αMAPK activity to expand thereby establishing the physiological milieu that characterizes the chronically inflamed SCQ 1, 2, 4. Consequently, it is this age-associated physiological milieu of chronic inflammation and the decline of FGFR1-Sprouty1 mediated regulation of P-p38αMAPK activity that plays a major role in the stabilized elevated P-p38αMAPK activity.
HS-FGF-2-FGFR1 and Sprouty 1 Synergistically Regulate the Homeostasis of P-p38αMAPK Activity in Juvenile MuSC
Our mechanism proposes that the autoinhibition of FGFR1 signaling and the Sprouty-1-mediated inhibition of the FGFR1 tyrosine kinase domains regulate the beneficial level of P-p38αMAPK activity in juvenile skeletal MuSC and that this coupled interaction is uncoupled in the aged MuSCs. This process of regulating FGFR1 signaling is supported by the observation that a) inhibition of FGFR1 signaling reduces the activity of P-p38α/βMAPK in juvenile MuSCs but not in aged MuSCs (uncoupled activity) 1, 2, 4. This enables age-associated elevation of P-p38αMAPK activity in response to sterile inflammation. Furthermore, inhibition of FGFR1 with SU5402 eliminates the coupled FGFR1 stimulation of p38α/βMAPK phosphorylation in juvenile MuSCs but has no effect on the phosphorylation of uncoupled p38α/βMAPK in aged MuSC [1]. These observations suggest that the elevated activity of P-p38α/βMAPK can proceed in the absence of HS-FGF-2-FGFR1 regulation and is due to and maintained by other signaling pathways, a likely candidate being chronic inflammation; b) the number of MuSCs in young mice can vary whereas the number of MuSCs in aged mice is not affected. This suggests an insensitivity of MuSCs from aged mice to over-sulfated HS-FGF-2 and supports our proposal that HS-FGF-2-FGFR1 controls the level of P-p38αMAPK activity in juvenile MuSC; that c) addition of FGF-2 to cultured young MuSC stimulates p38α/βMAPK phosphorylation in juvenile MuSCs but has no effect on p38α/βMAPK phosphorylation in aged MuSCs; d) p38α/βMAPK is insensitive to HS-FGF-2 due to the loss of function of FGFR1 signaling; e) loss of FGFR1 D1 or AB/linker domains by alternative splicing increases the signaling capacity of FGFR demonstrating that the splicing removed the sequences that control the coupling of FGFR1 to p38αMAPK [24]. These results suggest that there is a coupling of the HS-FGF-2-mediated stimulation of FGFR1 signaling that targets and activates p38α/βMAPK and highlights the insensitivity of p38α/βMAPK to over-sulfated HS-FGF-2 in aged MuSCs; f) ectopic activation of FGFR1 signaling via iFR1 and partial inhibition of p38α/βMAPK in MuSCs of aged mice restores asymmetric localization of P-p38α/βMAPK thus partially rescuing self-renewal in cultured cells [1,2].
The decline of MuSC self-renewal is linked to the increased transcription of the cell cycle inhibiting miRNAs, miR-1 and miR-133, in response to the sustained, elevated P-p38αMAPK activity and the simultaneous desensitization-mediated attenuation of an over-sulfated HS-FGFR1 complex (Figure 7). In our hypothesis, we propose that both loss of function of the HS-FGF-2-FGFR1 and gain of function of P-p38αMAPK is the underlying basis that promotes the development of SCQ in aged MuSC. This is consistent with our proposal that there is a coordinated, coupled rigorously controlled HS-FGF-2-FGFR1 regulation of the level of P-p38αMAPK activity that supports the replicative and regenerative capacity of juvenile tissue stem cells whereas in aged tissues attenuation by over-sulfated HS-mediated desensitization of the HS-FGF-2-FGFR1 complex enables the elevation of P-p38αMAPK activity thereby promoting a physiological milieu that supports the loss of asymmetry and replicative capacity of aged MuSCs.
Our mechanism proposes that the reversibility of the SCQ to a replicatively competent MuSC may be restored by the downregulation of over-sulfation of HS thereby regaining HS-FGF-2-FGFR1 functions and reactivation of the coupled control of the level of P-p38αMAPK activity. Our hypothesis thus proposes that the physiological consequences of desensitization-inactivation of HS-FGF-2-FGFR1 elevates P-p38αMAPK activity in chronically inflamed aged MuSCs and that these are physiological factors of the programming of the aging phenotype. Furthermore, an important component of our mechanism proposes that the elevated activity of P-p38αMAPK enhances the transcription of two microRNAs, miR-1 and miR-133, which are P-p38αMAPK-activated cell cycle inhibitors 38, 39.
Autoinhibition of FGFR1 Activity Plays a Protective Role by Regulating the Level of P-p38αMAPK Activity in Juvenile Skeletal MuSC
The mammalian FGFRs are transmembrane proteins whose extracellular regions consist of three immunoglobulin (Ig)-like ligand-binding domains (D1, D2 and D3), a transmembrane domain and an intracellular kinase domain (Figure 3, Figure 4). The D1, D2, and D3 extracellular domains are connected by a flexible linker (Figure 4; [43]). The D1-D2 linker is composed of a stretch of glutamate-, aspartate- and serine-rich sequences, i. e., the acid box (AB) [44]. The linker between D2 and D3 (D2-D3 region) is the site of FGF ligand binding and specificity [45]; and the heparan sulfate binding site (HS) which is located upstream in D2 (Figure 3, Figure 4). [46].
Removal of the AB/linker by alternative splicing produces an isoform that enhances the affinity of FGFR1 for HS-FGF-2 thereby releasing the coupled control of receptor signaling for its p38αMAPK target. Furthermore, it revealed that these linker sequences (AB) control receptor autoinhibition, and potentially the regulation of the level of p38αMAPK activity. This demonstrates that the level of P-p38αMAPK activity is linked to the AB/linker and strongly supports its regulation of P-p38αMAPK activity. We propose this as a potential protective mechanism that links the HS-FGF-2-FGFR1 control of p38αMAPK activity in juvenile MuSC, and allows for the maintenance of the beneficial levels of P-p38αMAPK activity that are critical to juvenile homeostasis 47, 48, 49, 50, 51, 52. The formation of the desensitized FGFR1 complex results in the uncoupling of the control of p38αMAPK activity. Furthermore, the removal or inactivation of this region supports the age-associated uncoupling by sterile inflammation-mediated elevation of P-p38αMAPK activity, which in turn stimulates miR-1/miR-133 and cell cycle arrest. The loss of the AB/linker by alternative RNA splicing in cancer cells suggest a mechanism that mediates the uncoupling of the linker-controlled regulation of P-p38αMAPK activity and consequently the loss of control of proliferation 53, 54, 55, 56.
Figure 3.The formation and stabilization of a 2:2:2 HS-FGF-2-FGFR1 signal transducing dimer. (a) The wild type HS-FGF-2-FGFR1 complex; (b) The extracellular complex consisting of the immunoglobulin domains, D1, D2, and D3; the FGF-2 ligand and bound heparan sulfate (HS). (c) The linker between immunoglobulin domains D2 and D3 affect the affinity and specificity of the receptor; (d) The intracellular tyrosine kinase domain (TK) that activates downstream targets, e.g p38αMAPK. (This figure is a modification of Figure 5 in DM Ornitz et al. Dev. Bio. 4, 215-266 (2015) with permission of the publisher
Figure 4.A schematic diagram of the extracellular FGFR Domains and the Intracellular Kinase Domains. The model shows the extracellular autoinhibition and ligand binding domains of FGFR1. The Acid Box or AB linker is involved in the process of autoinhibition and is localized between D1 and D2. In our model, direct inhibition of HS binding to FGFR1 attenuates the autoinhibitory process and is mediated by the AB linker (See Figure 7-Closed Model). On the other hand, the removal of the AB linker by alternative splicing eliminates the autoinhibitory function of the extracellular domain (D2). The heparan sulfate-binding site (HBS-Red) is in D2. The Ligand Binding Site is composed of the D2-D3 linker region and the D3 domain (GREEN). The heparan binding site (HBS: RED) is in D2 and the Heparan sulfate-FGF2 binding site (GREEN) is in D3. (This figure is a modification of Figure 2 in Mohammadi, M et al. Cytokine Growth Factor Rev. 16, 107-137 (2005) with permission of the publisher
The Role of HS-FGF-2-FGFR1-Signaling in Skeletal Muscle Regeneration and Reversal of the SCQ of Aged MuSCs.
As mice age the HS-FGF-2-FGFR1-p38 αMAPK-Axis of MuSCs undergoes a significant homeostatic alteration that involves the parallel loss of the HS-FGF-2 activation of FGFR1 signaling (desensitization); the over-sulfation of heparan; the development of a state of chronic inflammation (either sterile or exogenous), and the elevated level of P-p38αMAPK activity. A major consequence of these physiological changes induces the equal distribution of P-p38αMAPK to the MuSC daughter cells that results in the loss of asymmetric replication 1, 2, 3, 4 This gain of equal distribution of P-p38αMAPK activity establishes that both cells loose replicative capacity but retain capability of terminal differentiation [1,2,4,31]. Importantly, the increased level of P-p38αMAPK activity mediates the increased transcription of miR-1 and miR-133, both of which down regulate the cell cycle, thus promoting the SCQ (Figure 1). Secondly, the HS-FGF-2 activation of FGFR1 signaling is desensitized so that its rigorous control of P-p38αMAPK activity is uncoupled, thus enabling the increase in P-p38αMAPK activity to proceed in response to chronic inflammatory challenges. Furthermore, the failure of over-sulfated HS-FGF-2 to activate FGFR1 is an important characteristic of the overall physiological milieu of the aged skeletal muscle that contributes to the loss of replicative capacity of the MuSC. We propose that the loss of the rigorous homeostatic control of p38αMAPK activity by desensitization of the HS-FGF-2-FGFR1 complex enables the sterile inflammation-mediated elevation of the P-p38αMAPK activity to promote the loss of asymmetry and replicative capacity and the progression of the aging phenotype in response to sterile inflammation.
The Muscle Heparanome is Altered in Aged Skeletal Muscle: The Role of Over Sulfated Heparan Sulfate Modification in Aged Muscle
HS functions are linked to specific sulfation structural modifications that differentially activate HS-FGF-2-FGFR1 ligand-receptor binding 35, 57, 58, 58, 59, 60, 61. A major causative factor of the age-associated decrease in regenerative capacity of MuSC is attributed to the a) level of extracellular sulfated glycosaminoglycans (GAG), 1, 2, 13, 33, 34, 58, 62 and b) the pattern of heparan sulfate modifications. These physiological characteristics promote signaling alterations in aged MuSCs (Figure 5, Figure 6) 35, 58, 63. This involves an age-associated overall increase in 6-O-sulfation of HS as occurs in the quadriceps of aged mice (1 year and 2 year old) compared to young mice (3 mos old) 36. On the other hand, desulfation of the 2-O position in the HS chain promotes premature senescence in various cell lines, e.g., MCF7 and HDF cells 64 which is consistent with the cell type specificities of HS sulfation. Furthermore, the specificity of sulfation at the 2-O-position prevents the stimulation of FGFR1 signaling and senescence 62, 64. The 2-O position in HS thus plays an important role in preventing cellular senescence via the modulation of FGFR1 signaling. We propose that over-sulfated HS, a characteristic of aged MuSCs, dramatically reduces MuSC numbers, i.e., proliferative capacity, but does not affect differentiation, even at high concentrations 61. These differential responses are thus due to the increased age-associated HS levels, to differential sites of sulfation of HS oligosaccharides, and to the resultant structural changes caused by these modifications(Figure 5, Figure 6], this is attributed to cell cycle arrest promoted by over-sulfated heparan. This sequence of physiological events is consistent with the observation that there is an increase in 6-O-sulfation in aging muscle and that aged over-sulfated MuSCs are impaired [1, 2, 13, 33, 36]. Importantly our hypothesis is further supported by the observation that treatment with a specific over-sulfated HS mimetic results in the loss of MuSC proliferation [36, 61]. Thus, an increase in 6-O-sulfation, which is mimicked by 2-de-sulfated-N-acetylated heparin, also results in the loss of proliferation; this mimetic contains a greater proportion of 6-O-sulfation as a result of selective desulfation of the 2-O and N-positions (Figure 5, Figure 6]. Levels of 6-O-sulfation of endogenous HS increase with age as well as when MuSC-derived myoblasts exit the cell cycle to initiate terminal differentiation [36]. These observations support our proposal that age-associated loss of proliferative capacity of MuSC is linked to the desensitization of FGFR-1-signaling caused by the inability of over-sulfated HS-FGF-2 to bind to an oversulfated HS-FGFR-2 complex.This would explain the failure of an age-associated increase in over-sulfated HS-FGF-2 to stimulate MuSC proliferation. In general, the physiological activities of HS sulfation are consistent with the aging phenotype of MuSCs. This supports our proposal that modification of HS-sulfation patterns attenuate HS-mediated signaling and contribute to the loss of proliferative capacity.
The mechanism of activation of FGFR1 signaling involves the formation and stabilization of a 2:2 HS-FGF-2-FGFR1 signal transducing dimer (Figure 3, Figure 6c ; 44, 56). HS interacts with the heparan sulfate binding site (HBS) of the FGFR1 D2 domain and the HS-FGF-2 ligand binding site (D2-D3) (Figure 4, Figure 7). This enhances protein-protein interaction at the dimer interface thereby sustaining dimerization 65 which activates signaling via tyrosine phosphorylation of the intracellular kinase domains (Figure 6c; 66, 67). The binding of HS-FGF-2 creates the docking sites for recruitment and phosphorylation of downstream signaling substrates that mediate changes in gene expression 68. More specifically, identification of the structure that includes the sequence of monosaccharides and sites of sulfation of HS has shown that overall the minimal FGF HS binding sequence exhibits a high degree of FGF specificity. Thus, the specificity of the minimal FGF-2 HS binding sequence requires the 2-O-sulfation at the IdoA site combined with N-sulfation at GlcN as the key structural requirement of interaction (Figure 5, Figure 6; 58, 69). Furthermore, the importance of the specificity of HS sulfation for a particular FGF ligand is indicated by the specificity of the minimal FGF1 binding sequence, which is a structure, composed of 5-7 monosaccharides containing a critical trisulfated IdoA2S-GlcNS6S that is, however, inactive with FGF-2 (Figure 6). On the other hand, the inability of over-sulfated HS-FGF-2 to bind to the D2-D3 domain is a factor in the failure to elicit the FGFR1 dimerization required for its activation (Figure 4, Figure 6c, Figure 7). Thus, desensitization of FGFR1 may in part be due to the structural changes conferred upon a functional complex by an over-sulfated HS-FGF-2 thereby preventing its binding and activation of receptor phosphorylation of the intracellular kinase domains 66, 67. This orientation is consistent with the inability of over-sulfated HS-FGF-2 to bind to the D2-D3 domain and consequently the failure to elicit FGFR1 dimerization-activation (Figure 4, Figure 6, Figure 7). Thus, the importance of the position of sulfation is seen by the 6-O-sulfation of GlcN that is functional for FGF-1 but non-functional for FGF-2 binding 69. This demonstrates that alteration of these specific patterns of sulfation in aged MuSCs, as a consequence of over-sulfation of HS, is a major factor in the desensitization-dysregulation of the FGF-2-FGFR1 signaling complex.
Figure 5.Structural representations of heparan sulfate (HS) and heparin (Hp) and their binding motifs proposed for FGF-2 and FGF-1 (See also Figure 6) Heparan sulfate (HS) and Heparin (Hp) share a precursor disaccharide composed of glucuronic acid/iduronic (GlcA/IdoA) and glucosamine (GlcN). This precursor disaccharide is used to build up their backbone structure which is composed of D. Heparin (Hp) is composed of alternating 4-linked uronic acid and 4-linked a-glucosamine (GlcN) units with varying substitutions. The major disaccharide is predominantly composed of a disaccharide unit. Other disaccharides composed of non-sulfated iduronic acid (IdoA), glucuronic acid (GlcA), N-acetyl glucosamine (GlcNAc), N-sulfated glucosamine (GlcNS3S6S) also occur but at lower levels. Heparan sulfate (HS) is less modified than Hp and is mainly composed of alternating unsulfated GlcA and GlcNAC units. (This Figure is a modification of Figure 1A in Pomin, VH Biochimie 127, 214-226, (2016) with permission of the publisher.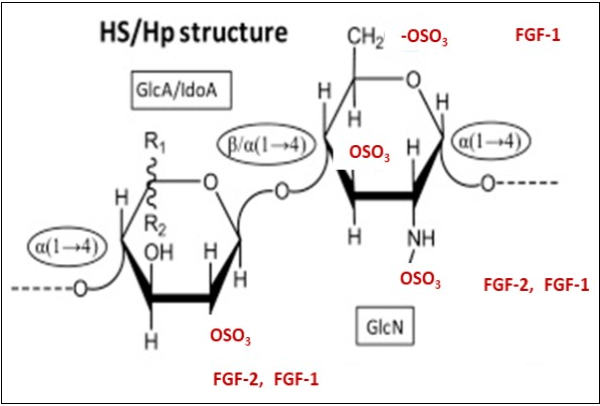
Figure 6.The (A) FGF-2 and (B) FGF-1 binding sites. Structural representations of Heparin (Hp) and Heparan sulfate (HS) and their binding motifs proposed for (A) FGF-2 and (B) FGF-1. (A) The FGF-2 binding site is proposed as the following pentasaccharide structure →4). (B) The FGF-1-binding site is proposed as the following pentasaccharide/heptasaccharide structure→4)-L-IdoA2S-α(1→4)-D-GlcNS6S-α(1 → 4)-L-IdoA2S-α(1→]. (C) Molecular model of FGF-2-FGFR1-Hp-tetrasaccharide [Pellegrini et al (2000); Schlessinger et al (2000)]. The model shows the surface of FGF-2, the D2 domain and the Hp tetrasacchride (stick model). O1 and O4 indicates the reducing and non-reducing ends and the 2:2;2 molecular ratio of the complex components The Hp ligands bind symmetrically on the canyon formed by the complex FGFR1 dimer-FGF-2-dimer In all panels the glycosidic bonds are indicated in ellipses whereas monosaccharide types are indicated in rectangles. Sulfation is highlighted in cyan. (This Figure is a modification of Figure 1B and 1C in Pomin VH Biochemie 127, 214-226 (2016) and Figure 4 in Schlessinger, J et al. Molecular Cell 6, 743-750 (2000) with permission from the publisher.![The (A) FGF-2 and (B) FGF-1 binding sites. Structural representations of Heparin (Hp) and Heparan sulfate (HS) and their binding motifs proposed for (A) FGF-2 and (B) FGF-1. (A) The FGF-2 binding site is proposed as the following pentasaccharide structure →4). (B) The FGF-1-binding site is proposed as the following pentasaccharide/heptasaccharide structure→4)-L-IdoA2S-α(1→4)-D-GlcNS6S-α(1 → 4)-L-IdoA2S-α(1→]. (C) Molecular model of FGF-2-FGFR1-Hp-tetrasaccharide [Pellegrini et al (2000); Schlessinger et al (2000)]. The model shows the surface of FGF-2, the D2 domain and the Hp tetrasacchride (stick model). O1 and O4 indicates the reducing and non-reducing ends and the 2:2;2 molecular ratio of the complex components The Hp ligands bind symmetrically on the canyon formed by the complex FGFR1 dimer-FGF-2-dimer In all panels the glycosidic bonds are indicated in ellipses whereas monosaccharide types are indicated in rectangles. Sulfation is highlighted in cyan. (This Figure is a modification of Figure 1B and 1C in Pomin VH Biochemie 127, 214-226 (2016) and Figure 4 in Schlessinger, J et al. Molecular Cell 6, 743-750 (2000) with permission from the publisher.](/article/1309/images/image6.png)
Figure 7.The heparan sulfate, FGF-2, FGFR1, interactions of the OPEN-CLOSE model that regulates the HS-FGF-2-FGFR1-p38αMAPK Axis in juvenile MuSCs and promote the desensitization-dysregulation of FGFR1 signaling in aged MuSCs. A, B, C: The CLOSED model in juvenile MuSC: The equilibrium in juvenile MuSC favors the OPEN function of HS-FGF2-FGFR-1-p38αMAPK axis. In this model there is a (A) controlled autoinhibition that prevents uncontrolled FGF signaling which controls the level of p38αMAPK activity; this also maintains beneficial levels of P-p38αMAPK activity and beneficial homeostatic levels of P-p38αMAPK activity. This supports a balanced myogenesis. In the aged MuSCs the equilibrium favors the desensitization-dysregulation of FGFR1 and inhibition of the HS-FGF-2-FGFR1-p38αMAPK Axis. The desensitization of. the Axis Heparan sulfate (HS) thus enforces the elevation and stabilization of levels of P-p38αMAPK activity. This physiological picture of the exogenous D1, D2, D3 FGFR1 domains that mediate the (D) ligand dependent dimerization needed for signal activation. In this model the level of autoinhibition of FGFR1 tightly regulates the level of coupled p38αMAPK signaling activity by (A) (a partial) steric autoinhibition of ligand binding to FGFR1; (B) Direct (partial) sulfoinhibition of HS binding to FGFR1; (C) Partial steric autoinhibition of receptor-receptor interaction. The levels of these interactions in the juvenile MuSCs regulate the beneficial, homeostatic levels of P-p38αMAPK (and other Axis targets) as depicted by the OPEN model. D, E, F: The OPEN model The equilibrium indicates that the OPEN model predominates thus promoting the progression of myogenesis. This establishes the level of P-p38αMAPK activity needed for the beneficial functions of the juvenile MuSCs (D, E, F). This physiological milieu promotes the replicative and regenerative capacity in myogenesis. This figure is a modification of Figure 6 in Kalinina J. al. Structure 20, 77-88 (2012) with permission from the publisher.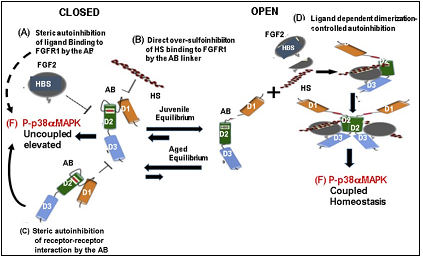
The OPEN-CLOSE Model of the HS-FGF-2-FGFR1-p38αMAPK Axis: Autoinhibition-Desensitization and Reversal of Replicative Capacity of Senescent MuSC.
Uncontrolled FGF signaling is associated with the development of human malignancies and other pathophysiological syndromes. The control of these FGF-linked pathophysiological characteristics is achieved by a self-regulatory mechanism, i.e., FGFR-autoinhibition that controls FGFR activity. This autoinhibition is mediated by the D1 domain and acid box (AB) containing linker (Figure 4, Figure 7). It serves as a defense mechanism that minimizes and controls the level of FGF-2 signaling that maintains the P-p38αMAPK activity required for efficient juvenile cell metabolic performance. This is critical for beneficial juvenile MuSC functions and homeostasis. Furthermore, with the progression of the MuSC aging phenotype, accumulation of over-sulfated HS-FGF-2 desensitizes FGFR1 signaling and loss of autoinhibitional control thereby enabling the elevation and stabilization of P-p38αMAPK activity in response to chronic sterile inflammation. We thus propose the OPEN-CLOSED model 24 as a mechanism by which the D1 and AB/linker mediate the molecular interactions that control the beneficial homeostatic levels of P-p38αMAPK activity and the regulation of proliferation of juvenile MuSCs.
The OPEN model shown in Figure 7D-F, provides a mechanism for the autoinhibition-mediated control of the level of coupled P-p38αMAPK activity essential for juvenile cell myogenesis, whereas age-associated modificationsdrivenby elevated over-sulfated HS-FGF2 and HS-modified receptor (HBS) results in the desensitization of FGFR1 and loss of the Axis regulatory function.
The mechanism by which D1 and AB/linker impose receptor autoinhibition is attributed to the “electrostatic interactions” of AB with D2 and intramolecular interactions of D1 with HS- FGF-2 ligand 24. We present the following mechanism of age-associated inactivation-desensitization of FGFR1 and the reactivation of the HS-FGF-2-FGFR1-p38αMAPK-Axis by the reprogramming of aged MuSC signaling to a juvenile phenotype (Figure 7D-F):
D1 prevents the binding of HS-FGF-2-ligand to D3 and HS-binding (HBS) interacting with D2-D3. This complex suppresses both HS-FGF-2 ligand (A) and HS-binding affinity of the receptor (B) 50). We propose that the failure of HS binding and HS-ligand-receptor interaction results in the partial autoinhibition that regulates the level of P-p38αMAPK activity. This occurs in the juvenile MuSCs and depends on the low level of HS over-sulfation. Alternatively, the desensitization of FGFR1 signaling in the aged MuSC is due to the age-associated increase of over-sulfated HS and shift in equilibrium (Figure 7B).
D. The Open Circle
In the juvenile MuSCs the negatively charged AB subregion of the AB/linker electrostatically engages the positively charged HBS site of D2 thereby enforcing a partial suppression of HS binding that promotes the autoinhibition of p38αMAPK (Figure 7D, E, F).
Surface electrostatic potential of the first monomeric HS-FGF-2-FGFR1 complex indicates that the HBS on D2 (Figure 4, Figure 7D) forms a contiguous positively charged surface (Figure 7C-F: 70 that promotes the regulation of p38αMAPK.
The electrostatic interactions of AB with D2 in turn promote intramolecular interactions of D1 with HS-FGF-2-ligand binding sites in the D2-D3 region to suppress ligand binding 47.
The AB/linker region plays an important role in FGFR1 autoinhibition and the coupled regulation of p38αMAPK levels in juvenile MuSCs (Figure 7D-F). This is a partial reversible desensitization of FGFR1.
The juvenile equilibrium controls the level of P-p38αMAPK activity via the level of HS-FGF-2-FGFR1 at equilibrium.
Desensitization of the HS-FGF-2-FGFR1 function is due to age-associated MuSC impairment, which involves the inhibition of the complex formation due to over-sulfation of HS-FGFR1 (Figure 7A-C).
In aged tissue the equilibrium shifts to the CLOSED FGFR1 structure. This desensitization decreases the Axis activation of p38αMAPK and enables the sterile inflammation-activation to elevate and stabilize P-p38α/β MAPK activity.
The conversion of FGFRs by alternative splicing produces isoforms whose altered functions include the loss of autoinhibition thereby resulting in an uncoupled increase of p38αMAPK activity. The alternatively spliced FGFRs regulate such normal processes as embryonic development and adult metabolism 24, 71, 72, 73 as well as cancer metabolism. We propose that autoinhibition of FGFRs, as mediated by either the interaction with over-sulfated HS-FGF-FGFR or alternatively spliced immunoglobulin (Ig) domain 1 (D1) and the acid box (AB)-containing linker, may play a role in the desensitization-dysregulation of FGFRs in aged MuSC. This is done by uncoupling and enabling elevated p38αMAPK activity to proceed thereby allowing the increased stabilized activity of this inflammatory pathway and its stimulation of cell cycle inhibiting miRNAs (Figure 1). We note here, however, that there is no evidence that alternative splicing occurs in aged MuSC.
Is the Age Associated FGFR1 Desensitization and Loss of Self-Renewal Associated with (a) Protein Modification or (b) Redox Regulation?
The increased P-p38αMAPK activity and its insensitivity to over-sulfated HS-FGF-2 in aged MuSCs 1, 2 is attributed to the loss of function of HS-FGF-2-FGFR1. However, other age-associated factors may contribute to the loss of function of HS-FGF-2-FGFR1 signaling such as: a) increased receptor post-translational modification, e.g., chronic inflammation-promoted oxidative modification of FGFR proteins; b) modification(s) that block signal transduction via the extracellular D1, D2, D3 domains or the FGFR-cytoplasmic kinase domain may alter the structure of the binding domains thereby resulting in desensitization and loss of function. The protein structure caused by disulfide linkages of D1, D2, or D3 (Figure 4) may be important for the maintenance of their functions which may be abrogated by their reduction to cysteines. These modifications may, for example, achieve a reversible sulfhydryl oxidation-reduction desensitization-reactivation of reduced D1, D2, D3 domains 74. Thus, HS-FGF-2-ligand mediated stimulation of FGFR1 may be desensitized by products of the metabolic environment of chronic inflammation.
On the other hand, there are 3 Gly residues, GOGCTG, also known as the Gly loop in the kinase domain that is a universally conserved signature motif among all protein kinases 75. This loop is directly involved in catalysis of ser/thr kinases, (cAMP dependent protein kinases 76 and FPS (a PTK; 77. The receptor kinases in FGF2 --> miR-29a all contain a cysteine residue (Cys 488 in FGFR1) which is a redox site in their kinase domain. The observation that FGFR1 is subject to redox regulation via the Cys residue in the Gly loop suggests that this may also be a regulatory event associated with the elevated redox state (sterile chronic inflammation) of the aged MuSCs. FGFR1 has been shown to form a disulfide linked dimer under oxidizing conditions, but mutation of Cys 488 does not completely abolish the dimerization suggesting that the oxidation of other residues may also be involved in dimerization. At the same time a symmetric dimer of FGFR2 has been reported 78 indicating that disulfide bridges can be part of the mechanism of dimerization and desensitization of the FGFRs. We thus propose that the SCQ of aged skeletal MuSCs may be susceptible to the oxidative milieu of chronic sterile inflammation (Figure 1).
Contributions of the Sprouty1 (spry1) and FGF2 --> miR-29a pathway: How do they fit into our Model?
Under normal juvenile skeletal muscle physiological conditions (homeostasis) the MuSCs, juvenile and aged, are in a state of reversible quiescence 33, 79, 80]; they also exhibit a negligible turnover. Only the juvenile MuSCs can enter the cell cycle when they are activated to proliferate in muscle repair and regeneration [80,81].
Monitoring of aged cycling MuSC identified two populations of cells, the most abundant of which showed an increase in breaking quiescence (as indicated by BudR incorporation). Very importantly, however, their potential for self-renewal and contribution to the progression of myogenesis is significantly curtailed 40, 79. These breakthrough senescent cells also have lower levels of Sprouty which decreases the level of inhibition of FGF-2 signaling and also serves as an important factor that promotes satellite cell quiescence 81. Thus, in the aged breakout MuSCs, although the inhibition of FGF-2 by Sprouty is reduced, the over-sulfated HS, the desensitization of FGFR1, and the elevated levels of P-p38αMAPK activity persist and maintain the ability to promote apoptosis rather than proliferation and myogenesis. This suggests that the uncoupled increased level of P-p38αMAPK activity promotes the p38αMAPK apoptotic pathway. It is interesting, therefore, that in the juvenile MuSC the level of Sprouty inhibitory activity combined with the controlled autoinhibition of FGFR1 appear to synergistically support the homeostatic level of beneficial activity of the HS-FGF-2-FGFR1-p38αMAPK Axis. This is in keeping with the Axis-coupled control of P-p38αMAPK activity, which is essential for the maintenance of a beneficial juvenile homeostasis. In the aged MuSC, however, although treatment with FGF-2 stimulates their exit from quiescence, these cells do not undergo self-renewal, but instead, exhibit an increase in cell death, a process that can be promoted by the elevated P-p38αMAPK activity.
The FGF2--> miR-29a Stimulation of Regenerative Proliferation.
TheFGF-2 mediated stimulation of regenerative myoblast proliferation is linked to the FGF-2 regulated activation of miR-29a that mediates basement membrane protein alterations in preparation for proliferation 82. Thus, the inhibition of major basement membrane structural proteins, e.g., Col4a1, Lamc 1, Nid2, and HSPg2, whose inhibition is associated with the initiation of MuSC regenerative-proliferation, in vivo, is regulated by miR-29a. Furthermore, evidence for the role of FGF-2 --> miR-29a activation in the stimulation of MuSC proliferation is supported by the observation that Tamoxifen-induced deletion of miR-29a in adult MuSC decreases the proliferation and formation of newly formed myofibers during both CTX-induced muscle injury and after a strong bout of exercise 82. This suggests that the desensitization of FGFR1 by over-sulfation of HS-FGF-2 in aged MuSCs has the potential to abrogate the FGF2 --> miR-29a pathway and its promotion of MuSC proliferation if this interaction is indeed linked to the influence of over-sulfation and the desensitization of FGF-2-FGFR1 signaling. On the other hand, if, in the aged MuSC FGF2 --> miR-29a is independent of an FGF-2-FGFR1 activation the over-sulfated HS-FGF-2 desensitized complex may independently down regulate the stimulation of miR-29a. Thus, the desensitized HS-FGF-2 may not be able to activate the miR-29a thereby failing to promote the dismantling of the basement membrane and initiation of proliferation. At the same time, these aged MuSC are activated and exit the quiescent state, but their self-renewal and contribution to the progression of myogenesis is significantly decreased. Since these MuSC take the pathway to apoptosis instead of proliferation-differentiation [33], we propose that the elevated P-p38αMAPK, which plays an important role in the stimulation of apoptosis, may be an important factor in promoting their cell death. We also propose that the desensitized FGFR1 may contribute to their apoptosis.
Although miR-29a stimulates myoblast proliferation it is not clear whether this process proceeds via the HS-FGF2-FGFR1-p38αMAPK Axis or whether it is a direct FGF2 --> miR-29a – proliferation pathway.
We have presented a mechanism by which HS-FGF-2-FGFR1 stimulates myoblast miR-1 and miR-133 transcription and downregulation of their targets, Sp1 and cyclin D1, which inhibits cell cycle progression. Furthermore, the lower levels of Sprouty inhibition of the receptor tyrosine phosphorylation and the FGF-2 stimulation of miR-29a in juvenile MuSCs support the breakout of senescent MuSCs and the initiation of their self-renewal and proliferation. We propose that these miRNAs, by directly controlling cell cycle inhibitor activity, and the integrity of the basement membrane synergistically regulate myogenic proliferation and differentiation. The desensitization-dysregulation of these synergistic interactions result in the failure of aged MuSCs to mediate MuSC self-renewal and regenerative proliferation.
Conclusions
We have proposed a molecular mechanism that links the activities of juvenile HS-FGF-2-FGFR1-p38αMAPK Axis and Sprouty1 to juvenile self renewal and the alterations of the Axis, and Sprouty1 activities, to the age-associated decline of self-renewal and regenerative capacity. Our mechanism proposes that the HS-FGF-2-FGFR1-p38αMAPK Axis and Sprouty1 activitiesare tightly regulated in juvenile skeletal MuSCs thereby supporting a beneficial homeostasis of the MuSC, while as aging progresses these satellite cells develop their Axis and Sprouty 1 senescent physiological characteristics. This enables the state of sterile inflammation to promote an elevated and sustained level of P-p38αMAPK activity in aged MuSC 1, 2, 4, 21, 22. This mechanism involves an impaired response of FGFR1 to over-sulfated HS-FGF-2-ligand that uncouples the FGFR1 regulation of p38αMAPK activity. These age - associated physiological developments synergistically promote the decline of self-renewal and regenerative capacity as follows: a) the development of desensitization-dysregulation of HS-FGF-2-FGFR1 signaling, that is a function of over-sulfated HS 1, 2 and b) desensitization uncouples the FGFR1 regulation of P-p38αMAPK activity while, c) the decreased inhibition of the kinase domain by Sprouty1 along with desensitization promotes the increased level of p38αMAPK activity by the state of sterile inflammation; d) the age-associated state of chronic inflammation can, in the absence of FGFR-controlled signaling, promote an elevated level of P-p38αMAPK activity 23, 24, 25, 26. We thus propose that the combined HS-FGF-2-FGFR1-p38αMAPK and Sprouty1 signaling are tightly linked processes that regulate the level of P-p38αMAPK activity, an event that is critical to the beneficial homeostasis and healthspan of juvenile MuSC; that the autoinhibitory processing of FGFR1 plays an important role in controlling the level of HS-FGF-2 signaling in juvenile MuSC thereby maintaining a beneficial level of p38αMAPK activity. These age-associated alterations result in an uncoupled desensitization of the FGFR1 regulation of P-p38αMAPK activity in aged satellite cells thereby enabling the chronic sterile inflammation and/or a response to exogenous toxins to promote and sustain an elevated P-p38αMAPK activity. An important consequence of the increased P-p38αMAPK activity involves the increased transcription of the cell cycle inhibitors, miR-1 and miR-133 37, 38. These miRNAs attenuate expression of the cell cycle regulators, SP1 and cyclin D1, resulting in a G1/S arrest i.e., the quiescence of the MuSCs (Figure 1).
The age-associated stimulation of the cell-cycle-inhibiting miRNAs, i.e., miR-1 and miR-133 is increased in response to the increased P-p38αMAPK activity. This promotes the SCQ of MuSC. At the same time, the desensitization-dysregulation of the HS-FGF-2-FGFR1 interaction uncouples its coordinated and rigorous control of P-p38αMAPK thus enabling an elevated, sterile, chronic endogenous and/or exogenous inflammation-mediated increased expression of P-p38α MAPK activity and miRNA transcription.
Our proposal, suggesting that HS-FGF-2-FGFR1 signaling regulates the level of P-p38αMAPK activity in juvenile MuSC and that this interaction is uncoupled in aged MuSCs, is experimentally supported by the observation that a) inhibition of FGFR1 signaling reduces P-p38α MAPK activity in juvenile MuSCs but not in aged MuSCs 1, 2, 4; b); inhibition of FGFR1 by SU5402 eliminates FGFR1 stimulation of P-p38α MAPK phosphorylation in juvenile MuSCs but has no effect on the phosphorylation in aged MuSCs 1; c) treatment of young MuSC in culture with FGF-2 stimulates P-p38α MAPK phosphorylation but has no effect upon aged MuSCs; d) addition of FGF-2 to young MuSCs in culture stimulates p38α/βMAPK phosphorylation but has no effect on p38α/βMAPK phosphorylation in aged MuSCs; e) p38α/βMAPK is insensitive to HS-FGF-2 due to the loss of function of FGFR1 signaling. This suggests that coupled HS-FGF-2 mediated stimulation of FGFR1 signaling targets and activates p38α/βMAPK; this highlights the insensitivity of p38α/βMAPK to over-sulfated HS-FGF-2 in aged MuSCs; f) ectopic activation of FGFR1 signaling and partial inhibition of p38α/βMAPK in MuSCs of aged mice restores asymmetric localization of p38α/βMAPK thus partially rescuing self-renewal in cultured cells [1, 2].
The extracellular matrix plays a major role in the promotion of juvenile skeletal muscle regeneration-proliferation and the age-associated loss of self-renewal. The FGF2 stimulation of miR-29a is another signaling pathway that stimulates the initiation of MuSC proliferation. This pathway involves the targeted downregulation of major basement membrane structural proteins of the skeletal muscle, which dismantles the basement membrane thereby allowing the transport of FGF-2 into MuSCs during its early phase of initiation of proliferation [82]. Thus, in aged MuSCs the expression and maintenance of higher levels of these basement membrane proteins favors MuSC quiescence as compared to their inhibition and dismantling of the basement membrane required for the initiation of proliferation of juvenile cells. The stimulation of FGF2 --> miR-29a thus promotes the dismantling of glycoproteins of the basement membrane that facilitates FGF-2 transport and activation of the FGF2-FGFR1 interaction and activation of the Axis.
The selective modifications of HS mimetics convincingly supports our proposal that the alteration of the levels or patterns of these modifications promote differential proliferation vs. differentiation [61]. These 3 specific key site modifications of the basic disaccaride, Gluc/IdoA and GlcnAc, i.e., R1, R2 and R3, (in Figure 6; [36], exhibit a high degree of specificity for their interaction with FGF ligand. The formation of additional 3-0 suflation provide evidence that selective sulfation modifications influence differentiation and proliferation [61]. Furthermore, varying levels of sulfation in specific positions and/or combinations that mimic the types of HS found in vivo, (sulfation in one, two and/or all three positions, HS sulfated in more than 3 positions, and over sulfation in which additional 3-O-sulfations occur in glucosamine and uronic acid), provide strong evidence that the activity of different HS mimetics are structure-specific and concentration dependent, and that almost all of the HS-mimetics produce a marked reduction in differentiation and an overall increase in numbers of heparan oversulfated cells. The exception to this general trend, the over-sulfated heparin mimetic, that did not reduce differentiation even at high concentrations, but dramatically reduced cell numbers attributed to the induction of cell cycle arrest [36]. This is consistent with the observation that sulfation levels of endogenous HS increase when MuSC derived myoblasts exit the cell cycle to differentiate. The transition of a replicative myoblast to a non-replicative myofiber is an excellent example of gaining/losing replicative vs non-replicative phenotypes. The metabolic characteristics of this event, i.e., the transition of a replicatively competent cell to a non-replicative fiber cell, is an excellent example of gaining/losing replicative capacity in terminal differentiation. Further studies showed that the HS mimetics did not affect myoblast fusion.
P-p38αMAPK actively participates as a regulator of myoblast proliferation-to-differentiation in juvenile MuSCs by inducing cell cycle withdrawal and the repression of myogenesis associated genes. Thus, in juvenile MuSCs, p38αMAPK exerts its myogenic functions, in part, by binding to and acting at specific chromatin sites that encompass a large set of myogenic gene promoters. This interaction facilitates myogenic gene activation and repression, i.e., the silencing of proliferation associated genes and activation of differentiation promoting genes [83]. Thus, by associating with chromatin, in juvenile cells, p38αMAPK uncovers classes of genes associated with the transition of myoblasts from proliferation-to-differentiation [83]. On the other hand the aged quiescent MuSC are exposed to elevated levels of p38αMAPK while the transmission from proliferation-to-differentiation is blocked. Thus, although the aged MuSC can be activated they proceed to enter the apoptotic pathway in response to the elevated p38αMAPK promotion of apoptosis [80, 84]. We thus propose that contrary to activation of the proliferation-to-differentiation taken by juvenile MuSC, which occurs under conditions of beneficial p38αMAPK homeostasis, the activated aged MuSCs response may be due to the state of chronic sterile inflammation, FGFR1 insensitivity and elevated levels of P-p38αMAPK.
Sirt1, a class III deacetylase, is another regulatory protein that mediates chromatin binding negative regulation of myogenesis. The negative response of Sirt1 may be promoted by the elevated levels of p38αMAPK. Thus, it appears that the consequences of elevated age-associated state of chronic inflammation may contribute to the loss of MuSC proliferative capacity.
p38αMAPK controls the transition of myoblasts from proliferating-to-differentiating stages. It also plays a key role in the inhibition of the differentiation program by promoting MEF2 transcriptional activity and MyoD/E47 heterodimer formation by direct phosphorylation of MEF2 and E47 [85]. p38αMAPK thus arises as a master kinase for reprogramming gene expression during the proliferation-to-differentiation transition of satellite cells, both in vitro and in vivo [86]. However, as noted above the age-associated, altered milieu supports the loss of replicative capacity and the fact that terminal differentiation can occur results in the decline of the MuSC pool level.
In juvenile MuSC, p38αMAPK is recruited to a large set of gene promoters. This facilitates the activation or repression of genes associated with myogenesis. On the other hand, the progression of the aging muscle phenotype promotes the decline of juvenile protective factors, i.e., proteins whose beneficial functions translate directly to the self-renewal of progenitor cells and the quality of life provided by a beneficial homeostasis. Thus, the characteristics of the aging phenotype (AP) are the consequences of the elevated levels of p38αMAPK activity. This is suggested by the downregulation of the level of p38αMAPK activity in the aged dominant negative p38MAPK mutant in which the appearance of multiple age-associated markers of aging are delayed. [18, 23]. Parabiosis, the pharmacological inhibition of p38α(AF/+)MAPK, and the genetic control i.e., downregulation of the level of p38αMAP in the dominant negative p38α(AF/+)MAPK mutant all indicate that the age-associated phenotype is associated with the elevated p38αMAPK promoted by the age-associated development of the state of chronic sterile inflammation. We conclude that the maintenance of low levels of p38αMAPK and a beneficial homeostasis (as in juvenile MuSC) may delay or attenuate the progression of the aging phenotype.
By using the dominant negative p38αMAPK mouse (DN- p38αAF/+) we demonstrated that the in vivo reduction of p38αMAPK activity in the gastrocnemius of the aged mutant (20 mos old) promoted the following changes: a) the age-associated decline of BuBR1, aldehyde dehydrogenases1A1 and mitochondrial ALDH2 were all delayed in the aged mutant; b) the age-associated increase of p16Ink4a and p19Arf tumor suppressor genes of the Cdkn2a locus was attenuated; c) the decreased level of hydroxynonenal protein adducts; the expression of COX2 and iNOS levels were attenuated; d) the delayed decline of senescent MuSC pool levels and, d) the loss of gastrocnemius muscle mass 23. These studies showed that the control of p38αMAPK activity promotes the delay of MuSC aging; and establishes the homeostasis of juvenile p38αMAPK activity 23, 87. We thus propose that p38αMAPK is crucial to the onset and progression of skeletal muscle differentiation. Moreover, it modulates the expression and/or activity of many of the genes involved in the transitional proliferation-to-differentiation and epigenetic regulation of myogenesis 87, 88.
Sirt1 is a member of the class III deacetylase family 89. It is regulated by the free concentration of NAD+ 90; and acetylated lysine 16 of histone H4 (H4K16ac] is its preferred histone substrate [91]. Thus, Sirt1 enzymology becomes a rapid biochemical system through which changes in the metabolic milieu can be converted into distinct epigenetic states and patterns of gene expression.
Sirt1 and p38αMAPK are chromatin interacting proteins that play a critical role in muscle differentiation. Sirt1 activity involves a negative effect on the activation of myogenesis 92. In this case the regulation of Sirt1 expression levels is dependent upon the decline in the NAD+/NADH ratio 96. Thus, the decline of Sirt1 levels that occurs in aged MuSC results in the differentiation of C2C12 and primary human skeletal myoblasts which is indicative of its effect on differentiation. Thus, in the aged MuSC both p38αMAPK and Sirt1 promote a decline in replicative potential. This is indicative of the role of Sirt1 in myofiber formation (terminal differentiation) in the myogenesis program. The regulatory Sirt1 - chromatin interaction occurs simultaneously with the p38αMAPK regulatory chromatin-associated processes. This suggests that the inflammation-mediated p38αMAPKαand metabolic-mediated Sirt1 regulated myogenesis demonstrate a co-ordinated participation and crosstalk interactions that promotes the proliferation-to-differentiation of juvenile MuSC.
The underlying molecular mechanisms responsible for the inhibition of myogenesis are attributed to the specific recruitment of Sirt1 to muscle regulatory regions on chromatin through its interaction with the MyoD/ACAF complex and the histone acetyltransferase. Sirt1 controls the activity of the myogenic factor, MEF2. This complex promotes the deactivation of histones on muscle specific enhancers and repression of the myogenic gene program. Furthermore, it has been shown that restricted glucose availability is one of the signals that inhibits muscle differentiation in a Sirt1 dependent manner. This involves the activation of AMPK, a sensor of cellular energy levels which results in enhanced expression of nicotinamide monoribosyl transferase (Nampt) that is the rate limiting enzyme in the biosynthetic pathway that synthesizes NAD+ from nicotinamide. The consequences of Nampt may thus alter the NAD+/NADH ratio thereby resulting in the down regulation of Srt1.
In addition to the negative role of Sirt1, studies with muscle cell precursors suggest that its enhanced expression promotes proliferation by inhibiting the expression of the cell cycle inhibitors, p21Waf/Cip and p21Kip94. Low oxygen growth conditions have also been identified as a factor that controls Sirt1 expression in MuSCs. Thus, p38αMAPK and Sirt1 appear to be central factors of the mechanism that controls the balance between muscle cell precursor proliferation and differentiation in response to environmental challenges.
Sirt1 is a member of the class III deacetylase family 89, 90. It is regulated by the free concentration of NAD+89, 90 and acetylated lysine 16 of histone H4 (H4K16ac] is its preferred histone substrate [91]. Thus, Sirt1 enzymology becomes a biochemical system through which metabolic changes can be converted into distinct epigenetic states and patterns of gene expression.
Following proliferation MuSC enter a quiescent state 84, 95, 96. In response to injury the activated quiescent juvenile MuSC enter the cell cycle, These cells are characterized by the presence of MyoD, a muscle specific transcription factor and give rise to proliferating muscle precursors which upon expression of myogenin proceed to repair the damaged muscle 79. During the transition from quiescent cells to activated cells the underlying changes in metabolism occur, i.e., a metabolic shift from fatty acid and pyruvate oxidation in quiescent cells to an increase in glycolysis and glutaminolysis in the activated proliferating cells [92]. This metabolic reprogramming is associated with a decrease in the intracellular NAD+/NADH ratio thereby reducing Sirt1-mediated deactivation of H4K16ac, and activation of the myogenic program. MuSC derived from mice with a muscle specific inactivation of the Sirt1 deacetylase domain (Sirt1mko) display increased H4K16ac and dysregulated activation of the myogenic program. In addition, the Sirt (mko) mice have reduced myofiber size and exhibit impaired muscle regeneration. The results of these experiments have demonstrated that MuSC undergo a switch from fatty acid and pyruvate oxidation to glycolysis which is associated with the NAD+/NADH ratio that mediates the down regulation of Sirt1 activity. This class III protein deacetylase uses NAD+ as a cofactor in such a way that its activity is modulated by the NAD+/NADH ratio.
Sirt1 is a relevant factor in chromatin remodeling by its histone deactivation and modulation. It regulates the activity of multiple transcription factors and co-regulators of cell metabolism, growth, differentiation, survival, apoptosis, inflammation and stress response 97. More specifically the linkage of Sirt1 activity to metabolic activity enables it to play a crucial role in muscle differentiation and metabolism that involves a negative effect on myogenesis 93 i.e., on the differentiation of primary human skeletal myoblasts. The complexity of myogenesis is therefore clearly seen in the crosstalk of the FGF2-FGFR1-p38αMAPK axis and Sirt1. Both of these signaling processes mediate chromatin modifications that synchronize the proliferation-to-differentiation process. The metabolic modifications, e.g., elevated levels of p38αMAPK and altered NAD+/NADH mediate major changes; their crosstalk results in the age-associated changes in MuSC proliferative capacity and differentiation.
Micro RNAs, many of which are associated with p38αMAPK, play a major regulatory role in the processes of skeletal muscle aging 98. This involves their interaction with p38αMAPK in the regulation of the cell cycle inhibitors, miR-1 and miR-133. Mi-RNAs are well established factors that promote the loss of replicative capacity of aged MuSC. p38αMAPK modulates the expression and/or activity of many of the miRNA players involved in the transcriptional and epigenetic regulation of myogenesis 87, 98
Two classes of molecule—non-coding small RNAs (miRNAs) and RNA binding proteins (RBPs) mediate the post-translational regulation of Sirt1. More than 16 miRNAs that are differentially expressed in aged skeletal muscle modulate Sirt1 expression one of which is miR-34a 98. PCR analyses have shown that the expression of miR-34a-5p and miR-19a are significantly elevated in senescent cells. PCR analyses show that the expression of miR-34a-5p (and mir-449-5p) are significantly higher in an elderly human cohort (mean age 82,4+-3.3 years compared to a young cohort (mean age 37.6 +,- 14.5 years) 98. Furthermore, the expression of key target genes in senescent skeletal muscle associated with Sirt1 and p38αMAPK signaling was significantly increased in sarcopenia patients [98]. In conclusion it was proposed that miR-19a and miR-34a play a regulatory role in skeletal muscle aging. The repression of levels of Sirt1 in cellular senescence involves the interaction of p38αMAPK, miR-19a, and miR-34a which are significant factors associated with human MuSC senescence.
These studies have shown that muscle aging is associated with an increase in 6-O-sulfation and it is well established that aged MuSCs are impaired. The increase in 6-O sulfation inhibits myogenesis by inhibiting myoblast differentiation without promoting proliferation, further support a key role for HS-mediated signaling in age-associated MuSC impairment.
Our mechanism proposes a synergistic interaction between HS-FGF2-FGFR-1-p38αMAPK, Sprouty1, miR-1, miR-133, miR-29a that mediate the juvenile proliferative activity vs the age-associated loss of proliferative capacity of the MuSC. Our hypothesis unifies the role of the extracellular niche and intracellular milieu in the stimulation of juvenile proliferation vs the age-associated decline of MuSC self-renewal and regenerative proliferation.
Abbreviations
FGF-2, Fibroblast Growth Factor-2; FGFR-1, Fibroblast Growth Factor Receptor-1; HS, Heparan sulfate; HS-FGF-2-FGFR1-p38αMAPK-Axis, Heparan sulfate Fibroblast Growth Factor -1 Fibroblast Growth Factor Receptor1-p38alpha MAPKinase Axis; IGA, Irreversible Growth Arrest; p38αMAPK, p38αMAPKinase; miR-1, microRNA-1; miR-133, microRNA-133; miR-29a, microRNA-29a; SCQ, senescent cell quiescence.
Ethics
NA
Consent for Publication
In progress
Availability of Data and Materials
References and figures presented in the text are available in PubMed
Funding
The Bertha and Robert Bucksch Foundation for Research on Aging
Author Contributions
JP is the sole author of this manuscript. JP wrote, edited and approved of this manuscript.
Acknowledgements
I am deeply, sincerely and gratefully indebted to the Bertha and Robert Buchsch Foundation for their many years of support of my research endeavors. I am especially grateful for the Bertha and Robert Bucksch Distingished Professorship in Research on Aging and its supprt of this article. I fondly acknowledge the personal friendship of Bertha Bucksch and her encouragement of my research on aging.
I sincerely thank Dr Krishna Mohan Sepuyu for his exhustive reiew of our maniscipt’
Author Information
Optional
References
- 1.Bernet J D, Doles J D, Hall J K, Kelly Tanaka K, Carter T A et al. (2014) p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nature medicine. 20(3), 265-71.
- 2.Cosgrove B D, Gilbert P M, Porpiglia E, Mourkioti F, Lee S P et al. (2014) Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nature medicine. 20(3), 255-64.
- 4.Blau H M, Cosgrove B D, Ho A T. (2015) The central role of muscle stem cells in regenerative failure with aging. Nature medicine. 21(8), 854-62.
- 5.Cheung T H, Rando T A. (2013) Molecular regulation of stem cell quiescence. Nature reviews Molecular cell biology. 14(6), 329-40.
- 6.Montarras D, L'Honore A, Buckingham M. (2013) Lying low but ready for action: the quiescent muscle satellite cell. , FEBS J 280(17), 4036-50.
- 7.Brack A S, Rando T A. (2007) Intrinsic changes and extrinsic influences of myogenic stem cell function during aging. Stem Cell Rev. 3(3), 226-37.
- 8.Garcia-Prat L, Sousa-Victor P, Munoz-Canoves P. (2013) Functional dysregulation of stem cells during aging: a focus on skeletal muscle stem cells. , FEBS J 280(17), 4051-62.
- 9.Conboy I M, Conboy M J, Smythe G M, Rando T A. (2003) Notch-mediated restoration of regenerative potential to aged muscle. , Science 302(5650), 1575-7.
- 10.Conboy I M, Conboy M J, Wagers A J, Girma E R, Weissman I L et al.Rejuvenation of aged progenitor cells by exposure to a young systemic environment. , Nature 433(7027), 760-4.
- 11.Carlson B M, Faulkner J A. (1996) The regeneration of noninnervated muscle grafts and marcaine-treated muscles in young and old rats. The journals of gerontology Series A, Biological sciences and medical sciences. 51(1), 43-9.
- 12.Carlson B M, Faulkner J A. (1989) Muscle transplantation between young and old rats: age of host determines recovery. , Am J Physiol.256(6Pt1): 262-6.
- 13.Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L et al.Geriatric muscle stem cells switch reversible quiescence into senescence. , Nature 506(7488), 316-21.
- 14.Gaur M, Ritner C, Sievers R, Pedersen A, Prasad M et al.Timed inhibition of p38MAPK directs accelerated differentiation of human embryonic stem cells into cardiomyocytes. , Cytotherapy 12(6), 807-17.
- 15.Geest C R, Coffer P J. (2009) MAPK signaling pathways in the regulation of hematopoiesis. , J Leukoc Biol 86(2), 237-50.
- 16.Ventura J J, Tenbaum S, Perdiguero E, Huth M, Guerra C et al. (2007) p38alpha MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nature genetics. 39(6), 750-8.
- 17.Molofsky A V, Slutsky S G, Joseph N M, He S, Pardal R et al. (2006) Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. , Nature 443(7110), 448-52.
- 18.Wong E S, X Le Guezennec, Demidov O N, Marshall N T, Wang S T et al. (2009) p38MAPK controls expression of multiple cell cycle inhibitors and islet proliferation with advancing age. Developmental cell. 17(1), 142-9.
- 19.Krishnamurthy J, Torrice C, Ramsey M R, Kovalev G I, Al-Regaiey K et al. (2004) Ink4a/Arf expression is a biomarker of aging. The Journal of clinical investigation. 114(9), 1299-307.
- 20.Hui L, Bakiri L, Stepniak E, Wagner E F. (2007) p38alpha: a suppressor of cell proliferation and tumorigenesis. Cell cycle. 6(20), 2429-33.
- 21.Knight J D, Kothary R. (2011) The myogenic kinome: protein kinases critical to mammalian skeletal myogenesis. Skelet Muscle. 1, 29.
- 22.Pawlikowski B, Vogler T O, Gadek K, Olwin B B. (2017) Regulation of skeletal muscle stem cells by fibroblast growth factors. Dev Dyn. 246(5), 359-67.
- 23.Papaconstantinou J, Wang C Z, Zhang M, Yang S, Deford J et al.Attenuation of p38alpha MAPK stress response signaling delays the in vivo aging of skeletal muscle myofibers and progenitor cells. , Aging 7(9), 718-33.
- 24.Kalinina J, Dutta K, Ilghari D, Beenken A, Goetz R et al.The alternatively spliced acid box region plays a key role in FGF receptor autoinhibition. , Structure 20(1), 77-88.
- 25.Hsieh C C, Papaconstantinou J. (2002) The effect of aging on p38 signaling pathway activity in the mouse liver and in response to ROS generated by 3-nitropropionic acid. Mechanisms of ageing and development. 123(11), 1423-35.
- 26.Papaconstantinou J, Hsieh C C. (2010) Activation of senescence and aging characteristics by mitochondrially generated ROS: how are they linked? Cell Cycle. 9(19), 3831-3.
- 27.Calder P C, Bosco N, Bourdet-Sicard R, Capuron L, Delzenne N et al. (2017) Health relevance of the modification of low grade inflammation in ageing (inflammageing) and the role of nutrition. Ageing research reviews. 40, 95-119.
- 28.Lescai F, Conti L, Bartolozzi M, Ramazzotti G, Mazzi M et al. (2005) Genotype of inflammatory cytokines in limbal stem cell graft in Italian patients. Biochemical and biophysical research communications. 332(1), 95-100.
- 29.Freund A, Patil C K, Campisi J. (2011) p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. The EMBO journal. 30(8), 1536-48.
- 30.Perdiguero E, Ruiz-Bonilla V, Gresh L, Hui L, Ballestar E et al. (2007) Genetic analysis of p38 MAP kinases in myogenesis: fundamental role of p38alpha in abrogating myoblast proliferation. The EMBO journal. 26(5), 1245-56.
- 31.Troy A, Cadwallader A B, Fedorov Y, Tyner K, Tanaka K K et al. (2012) Coordination of satellite cell activation and self-renewal by Par-complex-dependent asymmetric activation of p38alpha/beta MAPK. Cell stem cell. 11(4), 541-53.
- 32.Puri P L, Wu Z, Zhang P, Wood L D, Bhakta K S et al. (2000) Induction of terminal differentiation by constitutive activation of p38 MAP kinase in human rhabdomyosarcoma cells. Genes Dev. 14(5), 574-84.
- 33.Chakkalakal J V, Jones K M, Basson M A, Brack A S.The aged niche disrupts muscle stem cell quiescence. , Nature 490(7420), 355-60.
- 34.Li J, Han S, Cousin W, Conboy I M. (2015) Age-specific functional epigenetic changes in p21 and p16 in injury-activated satellite cells. Stem cells. 33(3), 951-61.
- 35.Pye D A, Vives R R, Turnbull J E, Hyde P, Gallagher J T. (1998) Heparan sulfate oligosaccharides require 6-O-sulfation for promotion of basic fibroblast growth factor mitogenic activity. The Journal of biological chemistry. 273(36), 22936-42.
- 36.Ghadiali R S, Guimond S E, Turnbull J E, Pisconti A. (2017) Dynamic changes in heparan sulfate during muscle differentiation and ageing regulate myoblast cell fate and FGF2 signalling. Matrix Biol. 59, 54-68.
- 37.Engel F B, Schebesta M, Duong M T, Lu G, Ren S et al. (2005) p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 1175-87.
- 38.Zhang D, Li X, Chen C, Li Y, Zhao L et al. (2012) Attenuation of p38-mediated miR-1/133 expression facilitates myoblast proliferation during the early stage of muscle regeneration. PloS one. 7-7.
- 39.Zhang Y, Zhang L, Chu W, Wang B, Zhang J et al. (2010) Tanshinone IIA inhibits miR-1 expression through p38 MAPK signal pathway in post-infarction rat cardiomyocytes. Cell Physiol Biochem. 26(6), 991-8.
- 40.Abou-Khalil R, Brack A S. (2010) Muscle stem cells and reversible quiescence: the role of sprouty. Cell cycle. 9(13), 2575-80.
- 41.Perdiguero E, Ruiz-Bonilla V, Serrano A L, Munoz-Canoves P. (2007) Genetic deficiency of p38alpha reveals its critical role in myoblast cell cycle exit: the p38alpha-JNK connection. Cell cycle. 6(11), 1298-303.
- 42.Milasincic D J, Calera M R, Farmer S R, Pilch P F. (1996) Stimulation of C2C12 myoblast growth by basic fibroblast growth factor and insulin-like growth factor 1 can occur via mitogen-activated protein kinase-dependent and -independent pathways. Molecular and cellular biology. 16(11), 5964-73.
- 43.Plotnikov A N, Hubbard S R, Schlessinger J, Mohammadi M. (2000) Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. , Cell 101(4), 413-24.
- 44.Johnson B A. (2004) Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods in molecular biology. 278, 313-52.
- 45.Mohammadi M, Olsen S K, Ibrahimi O A. (2005) Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 16(2), 107-37.
- 46.Schlessinger J, Plotnikov A N, Ibrahimi O A, Eliseenkova A V, Yeh B K et al. (2000) Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Molecular cell. 6(3), 743-50.
- 47.Olsen S K, Ibrahimi O A, Raucci A, Zhang F, Eliseenkova A V et al. (2004) Insights into the molecular basis for fibroblast growth factor receptor autoinhibition and ligand-binding promiscuity. Proceedings of the National Academy of Sciences of the United States of America 101(4), 935-40.
- 48.Shi E, Kan M, Xu J, Wang F, Hou J et al. (1993) Control of fibroblast growth factor receptor kinase signal transduction by heterodimerization of combinatorial splice variants. Molecular and cellular biology. 13(7), 3907-18.
- 49.Shimizu A, Tada K, Shukunami C, Hiraki Y, Kurokawa T et al. (2001) A novel alternatively spliced fibroblast growth factor receptor 3 isoform lacking the acid box domain is expressed during chondrogenic differentiation of ATDC5 cells. The Journal of biological chemistry. 276(14), 11031-40.
- 50.Wang F, Kan M, Yan G, Xu J, McKeehan W L. (1995) Alternately spliced NH2-terminal immunoglobulin-like Loop I in the ectodomain of the fibroblast growth factor (FGF) receptor 1 lowers affinity for both heparin and FGF-1. The Journal of biological chemistry. 270(17), 10231-5.
- 51.Xu X, Weinstein M, Li C, Naski M, Cohen R I et al. (1998) Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction. Development. 125(4), 753-65.
- 52.Roghani M, Moscatelli D. (2007) Prostate cells express two isoforms of fibroblast growth factor receptor 1 with different affinities for fibroblast growth factor-2. Prostate. 67(2), 115-24.
- 53.Kobrin M S, Yamanaka Y, Friess H, Lopez M E, Korc M. (1993) Aberrant expression of type I fibroblast growth factor receptor in human pancreatic adenocarcinomas. Cancer research. 53(20), 4741-4.
- 54.Mansson P E, Adams P, Kan M, McKeehan W L. (1989) Heparin-binding growth factor gene expression and receptor characteristics in normal rat prostate and two transplantable rat prostate tumors. Cancer research. 49(9), 2485-94.
- 55.Tomlinson D C, Knowles M A. (2010) Altered splicing of FGFR1 is associated with high tumor grade and stage and leads to increased sensitivity to FGF1 in bladder cancer. , Am J Pathol 177(5), 2379-86.
- 56.Yamaguchi F, Saya H, Bruner J M, Morrison R S. (1994) Differential expression of two fibroblast growth factor-receptor genes is associated with malignant progression in human astrocytomas. Proceedings of the National Academy of Sciences of the United States of America 91(2), 484-8.
- 57.Rapraeger A C, Krufka A, Olwin B B. (1991) Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. , Science 252(5013), 1705-8.
- 58.Pomin V H.Paradigms in the structural biology of the mitogenic ternary complex FGF: FGFR: heparin. , Biochimie 2016, 214-26.
- 59.Kan M, Wu X, Wang F, McKeehan W L. (1999) Specificity for fibroblast growth factors determined by heparan sulfate in a binary complex with the receptor kinase. The Journal of biological chemistry. 274(22), 15947-52.
- 60.Reich-Slotky R, Bonneh-Barkay D, Shaoul E, Bluma B, Svahn C M et al. (1994) Differential effect of cell-associated heparan sulfates on the binding of keratinocyte growth factor (KGF) and acidic fibroblast growth factor to the KGF receptor. The Journal of biological chemistry. 269(51), 32279-85.
- 61.Yates E A, Guimond S E, Turnbull J E. (2004) Highly diverse heparan sulfate analogue libraries: providing access to expanded areas of sequence space for bioactivity screening. J Med Chem. 47(1), 277-80.
- 62.Guimond S E, Turnbull J E. (1999) Fibroblast growth factor receptor signalling is dictated by specific heparan sulphate saccharides. Curr Biol. 9(22), 1343-6.
- 63.Zhu H, Duchesne L, Rudland P S, Fernig D G. (2010) The heparan sulfate co-receptor and the concentration of fibroblast growth factor-2 independently elicit different signalling patterns from the fibroblast growth factor receptor. Cell Commun Signal. 8, 14.
- 64.Jung S H, Lee H C, Yu D M, Kim B C, Park S M et al. (2016) Heparan sulfation is essential for the prevention of cellular senescence. Cell Death Differ. 23(3), 417-29.
- 65.Mohammadi M, Olsen S K, Goetz R. (2005) A protein canyon in the FGF-FGF receptor dimer selects from an a la carte menu of heparan sulfate motifs. Curr Opin Struct Biol. 15(5), 506-16.
- 66.Chen H, Xu C F, Ma J, Eliseenkova A V, Li W et al. (2008) A crystallographic snapshot of tyrosine trans-phosphorylation in action. Proceedings of the National Academy of Sciences of the United States of America 105(50), 19660-5.
- 67.Mohammadi M, Dikic I, Sorokin A, Burgess W H, Jaye M et al. (1996) Identification of six novel autophosphorylation sites on fibroblast growth factor receptor 1 and elucidation of their importance in receptor activation and signal transduction. Molecular and cellular biology. 16(3), 977-89.
- 68.Eswarakumar V P, Lax I, Schlessinger J. (2005) Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 16(2), 139-49.
- 69.Maccarana M, Casu B, Lindahl U. (1993) Minimal sequence in heparin/heparan sulfate required for binding of basic fibroblast growth factor. The Journal of biological chemistry. 268(32), 23898-905.
- 70.Plotnikov A N, Schlessinger J, Hubbard S R, Mohammadi M. (1999) Structural basis for FGF receptor dimerization and activation. , Cell 98(5), 641-50.
- 71.Goldfarb M. (2005) Fibroblast growth factor homologous factors: evolution, structure, and function. Cytokine Growth Factor Rev. 16(2), 215-20.
- 72.Itoh N, Ornitz D M. (2011) Fibroblast growth factors: from molecular evolution to roles in development, metabolism and. 149(2), 121-30.
- 73.Kuro-o M. (2008) Endocrine FGFs and Klothos: emerging concepts. Trends Endocrinol Metab. 19(7), 239-45.
- 74.Caccia P, Nitti G, Cletini O, Pucci P, Ruoppolo M et al. (1992) Stabilization of recombinant human basic fibroblast growth factor by chemical modifications of cysteine residues. , EurJBiochem 204(2), 649-55.
- 75.Kemble D J, Sun G. (2009) Direct and specific inactivation of protein tyrosine kinases in the Src and FGFR families by reversible cysteine oxidation. Proceedings of the National Academy of Sciences of the United States of America 106(13), 5070-5.
- 76.Grant B D, Hemmer W, Tsigelny I, Adams J A, Taylor S S. (1998) Kinetic analyses of mutations in the glycine-rich loop of cAMP-dependent protein kinase. Biochemistry. 37(21), 7708-15.
- 77.Hirai T J, Tsigelny I, Adams J A. (2000) Catalytic assessment of the glycine-rich loop of the v-Fps oncoprotein using site-directed mutagenesis. Biochemistry. 39(43), 13276-84.
- 78.Chen H, Ma J, Li W, Eliseenkova A V, Xu C et al. (2007) A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Molecular cell. 27(5), 717-30.
- 80.Rodgers J T, Rando T A. (2012) Sprouting a new take on stem cell aging. The EMBO journal. 31(21), 4103-5.
- 81.Shea K L, Xiang W, LaPorta V S, Licht J D, Keller C et al. (2010) Sprouty1 regulates reversible quiescence of a self-renewing adult muscle stem cell pool during regeneration. Cell stem cell. 6(2), 117-29.
- 82.Galimov A, Merry T L, Luca E, Rushing E J, Mizbani A et al. (2016) . MicroRNA-29a in Adult Muscle Stem Cells Controls Skeletal Muscle Regeneration During Injury and Exercise Downstream of Fibroblast Growth Factor-2. Stem cells 34(3), 768-80.
- 83.Chen S E, Jin B, Li Y P. (2007) TNF-alpha regulates myogenesis and muscle regeneration by activating p38 MAPK. , Am J Physiol Cell Physiol 292(5), 1660-71.
- 84.Tajbakhsh S. (2009) Skeletal muscle stem cells in developmental versus regenerative myogenesis. , J Intern Med 266(4), 372-89.
- 85.Lluis F, Ballestar E, Suelves M, Esteller M, Munoz-Canoves P. (2005) . E47 phosphorylation by p38 MAPK promotes MyoD/E47 association and muscle-specific 24(5), 974-84.
- 86.Brien P, Pugazhendhi D, Woodhouse S, Oxley D, Pell J M. (2013) p38alpha MAPK regulates adult muscle stem cell fate by restricting progenitor proliferation during postnatal growth and repair. Stem cells. 31(8), 1597-610.
- 87.Segales J, Perdiguero E, Munoz-Canoves P. (2016) Regulation of Muscle Stem Cell Functions: A Focus on the p38 MAPK Signaling Pathway. Front Cell Dev Biol. 4, 91.
- 88.Zhang Y, Zhang G, Liu Y, Chen R, Zhao D et al. (2018) Leading to Immune Tolerogenic DCs for Preventing Alloimmune Rejection in Heart Transplantation. Front Immunol. GDF15 Regulates Malat-1 Circular RNA and Inactivates NFkappaB Signaling 9, 2407.
- 89.Michan S, Sinclair D. (2007) Sirtuins in mammals: insights into their biological function. , Biochem 404(1), 1-13.
- 90.Imai S I, Guarente L. (2016) It takes two to tango: NAD(+) and sirtuins in aging/longevity control. NPJ Aging Mech Dis. 2, 16017.
- 91.Vaquero A, Sternglanz R, Reinberg D. (2007) NAD+-dependent deacetylation of H4 lysine 16 by class III HDACs. Oncogene. 26(37), 5505-20.
- 92.Ryall J G, Dell'Orso S, Derfoul A, Juan A, Zare H et al. (2015) The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell stem cell. 16(2), 171-83.
- 93.Fulco M, Schiltz R L, Iezzi S, King M T, Zhao P et al. (2003) Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Molecular cell. 12(1), 51-62.
- 94.Rathbone C R, Booth F W, Lees S J. (2009) Sirt1 increases skeletal muscle precursor cell proliferation. , Eur J Cell Biol 88(1), 35-44.
- 95.Kim H J, Moon H S, Delhom C D, Zeng L, Fang D D. (2013) Molecular markers associated with the immature fiber (im) gene affecting the degree of fiber cell wall thickening in cotton (Gossypium hirsutum L.). Theor Appl Genet. 126(1), 23-31.
- 96.Pallafacchina G, Francois S, Regnault B, Czarny B, Dive V et al. (2010) An adult tissue-specific stem cell in its niche: a gene profiling analysis of in vivo quiescent and activated muscle satellite cells. Stem Cell Res. 4(2), 77-91.